
 企业邮箱
企业邮箱 企业OA
企业OA整理丨麦科奥特毒理学部高级总监 @ 张曾源编撰
编辑丨质量保证 (QA) 专员 @ 温朝霞
编辑丨早期开发部靶点研究员 @ 张家宁
编辑丨人力资源部行政主管 @ 李娜娜

如何在中国药企建立电子通用技术文档 (Electronic common technical document, eCTD) 管理系统, 使之有效地应用在各国的医事法规监管机构 (Regulatory authorities) 部门上申请研究性新药(Investigational new drug, IND)的注册,借此完成临床批文 (Clinical trial application, CTA) 的申报及审批,这个已成为现今中国药企必须面对的一个重要且急迫性的议题。
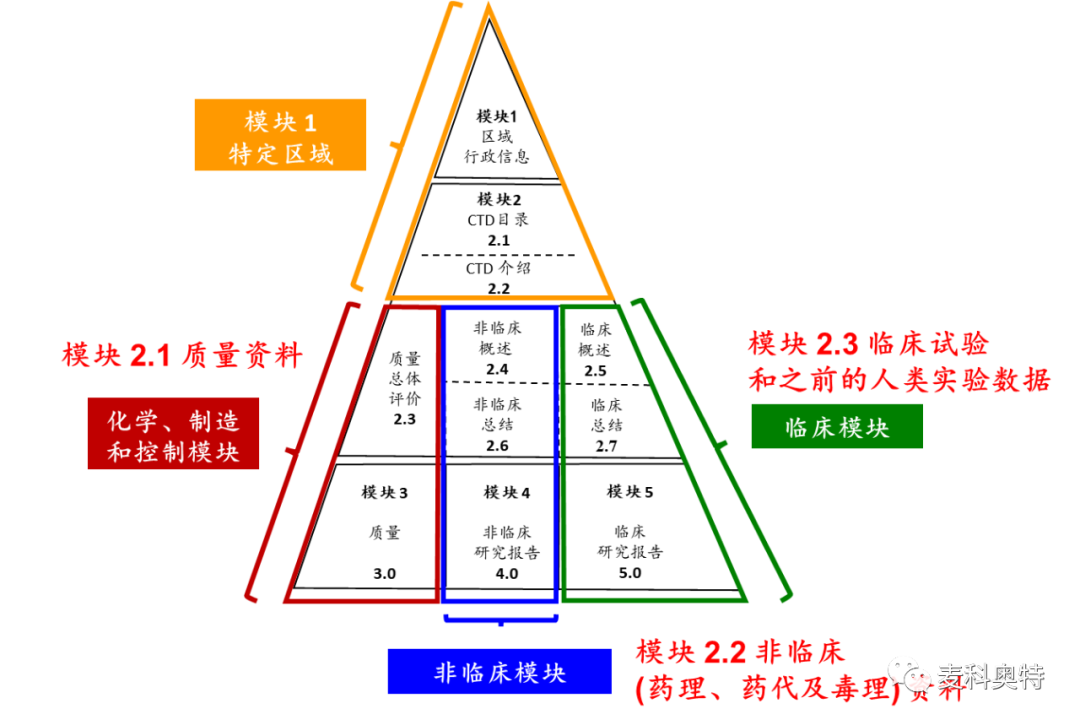
图 1 ICH 的通用技术文件 (CTD) 结构用于 IND 申报
图中模块里黑色字部分是属于在中国 (NMPA) 及美国 (FDA) 申报的章节,红色字部分是属于欧洲 (EMA) 申报的章节。
eCTD 是由国际协调委员会 (International Committee for Harmonization, ICH) 制定发布的规范,药品注册申报时申报企业按照规范编制结构化的药品电子申报资料,医事法规监管机构以电子化形式受理和审评,实现申报资料由递交到审评的规范化、标准化、信息化和电子化。 eCTD 是使用可扩展标记语言 (Extensible markup language, XML) 为基础,规范和展示CTD文件的名称和架构。而 XML 是一种标记语言,XML是从标准通用标记语言 (Standard generalized markup language, SGML) 中简化修改出来的,这种语言的优点为: i) 利用可用于处理的技术在文档群 (或数据库) 中允许标记,ii) 在建立的文档中对于已有标记保有严格定义程序, 可以有效的查询/管理所有资料库的资料。因为有以上的优点,加上 eCTD 是以XML为基础,管理/展示CTD文件的名称和架构,这种资讯管理架构/技术易于将eCTD 规范的药品申报资料以电子化形式进行组织、传输和呈现,有助于实现药品全生命周期的管理,这也就是为什么全球大多数的医事法规监管机构使用这个格式用于 IND 的申报、管理及审批。
下面就以中国国家药品监督管理局 (National Medical Products Administration, NMPA)、美国食品和药物管理局 (Food and Drug Administration, FDA) 与欧洲药品管理局 (European Medicines Agency, EMA) 的eCTD的架构做一个介绍。在中国及美国医事法规监管机构 (NMPA and FDA) CTD 包含有五个模块 (Modules)。其中模块 1 是用于新型药物的一般资讯他与/或特定于地区的药物申报专属资料,模块 2.3 与3 是介绍 “化学、制造和控制 (Chemistry, manufacturing, and. control, CMC)” 资料,模块 2.4、2.6 与4 是包含 ”非临床 (Nonclinical, NC)” 资料,模块 2.5、2.7 与模块 5 旨在说明 ”临床 (Clinical)” 资料。而在欧盟EMA 新型药物注册研究性药品档案 (Investigational medicinal product dossier, IMPD) 的模块是分为: 模块2.1质量数据 (CMC)、模块2.2非临床 (药理、药代及毒理) 资料,以及模块 2.3 临床试验和之前的人类实验数据 (图 1)。
图中模块里黑色字部分是属于在中国 (NMPA) 及美国 (FDA) 申报的章节,红色字部分是属于欧洲 (EMA) 申报的章节。
以美国FDA 为例,从 2008 年起,FDA 开始鼓励药企/申报方 (Applicants/sponsors) 以 eCTD 格式提交相关新药申请材料,以加速新药送件及审批流程。从 2010 年开始将新药上市注册申请、生物制品许可申请纳入 eCTD 观念里范围。自 2015 年 FDA 正式宣布,到 2017 年中旬前新药药物的申请将被要求采用 eCTD 提交,也就是强制要求药企申报方一定要使用 eCTD 提交新药审批文件,并在这一期限之后,FDA 将不再受理纸质文件的新药申请。到目前为止,全球已经有超过 35 个国家和地区的医事法规监管机构已经采用了 eCTD 格式递交注册文件,现在执行的标准依据是 2008 年发布的国际医药法规协和会有关电子用技术文件之相关规范 (Version 3.2.2)。
中国药政为了与接轨国际新药药物审批的标准,在近10年所执行的步骤如下: i) 中国国务院于 2015 年 8 月发布 “关于改革药物医疗器械评审审批制度的意见”,ii) 中国国家药品监督管理局(NMPA)于 2017 年 5月公布 “药品电子通用技术文档结构” (征求意见稿) 和 “化学仿制药电子通用技术文档申报指导原则” (征求意见稿),iii) 在后同年6 月加入 ICH,iv) 于 2018 年 7 月完成 eCTD 后台管理系统(验证,审阅,文档管理)招标, v) 并于 2019 年相继发布了技术规范、验证标准和申报指南等规范, vi) 于2020年5月 CDE 发布药物临床试验数据递交指导原则,明确 eCTD申报模块的部分,v) 在2020年9月CDE再次发布 “技能规范” 、 “验证标准”, 及 “eCTD 申报指南” 征求意见稿。再一次验证了中国新药格式递交注册文件格式要与国际标准接轨的决心。
那为何要使用eCTD来规范新药格式递交注册文件格式? 因为eCTD 带来的价值有: a) 在于促进药品国内外同步申报,便于申报资料的重复利用,同时有利于全球研发申报,并加快药品全球同步上市,缩短文件在重新编写时造成中国药企时间及金钱上的损失, b) 在提高药品申报成功率方面来看,因为中国是使用 eCTD 技术规范和申报指南,中国药企通过eCTD申报软件,加快申报资料准备,有助自查和修改,及时更新法规变化,可以大幅提高申报成功率, c) 因为申报文件符合乎国际 eCTD 技术规范,医事法规监管机构使用电子化审评方式,大幅度提高审评效率,缩短审评周期,4) 并通过高效率的申报全生命周期 (Lifecycle) 管理,实现对提交文档的历史追踪和版本管理,避免错报重报, 及 5) 在这一套系统下,有利于现场检查和核查,并可缩短新药上市时间,造福研发药物研发企业及受惠的用药病患。
eCTD 的架构
任何中国药企在中国和/或美国医事法规监管机构使用 eCTD新药申请,经由商业化的 eCTD 编辑软体可以输出所提交的全套申报资料,所有输出文件及档案夹必须包括有: i) 模块 1-5 (Modules 1-5) 的档案夹, ii) 2个骨架文件 (.xml文件), iii) 1个文档类型定义相关文件档案夹 (Document Type Definition, DTD;包括有: 元素 (Elements)、属性(Attribute)、实体(Entities)及注释(Comments))及Style的资料会放置在 util文件档案夹内,及 iv) 1个 XML档案 (index.xml) 及 1个 Message-digest algorithm (MD5) 档案 (index-md5.txt) (图 2)。
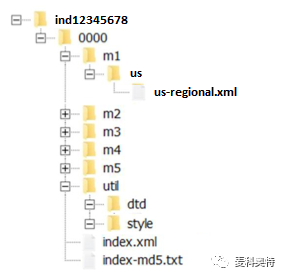
图 2 借由eCTD技术提交 ind12345678的全套申报资料输出档案夹的结构示范图
模块 1 最主要要展示的是行政信息和处方信息 (Administrative information and prescribing information): 该模块包含每个区域唯一的管理信息,其中区域指导将提供有关如何提供行政表格和详细处方的具体说明信息 (图 3)。
模块 2 总结 (Summaries): 模块 2 的档案夹名称应为 m2,此模块中最主要是介绍 CMC、临床前以及临床的主要内容摘要,此模块的档案夹应命名如图 4 所显示,其档案夹名称可以进一步减少或以最小化档案夹名称来解决档案名称长度的问题。

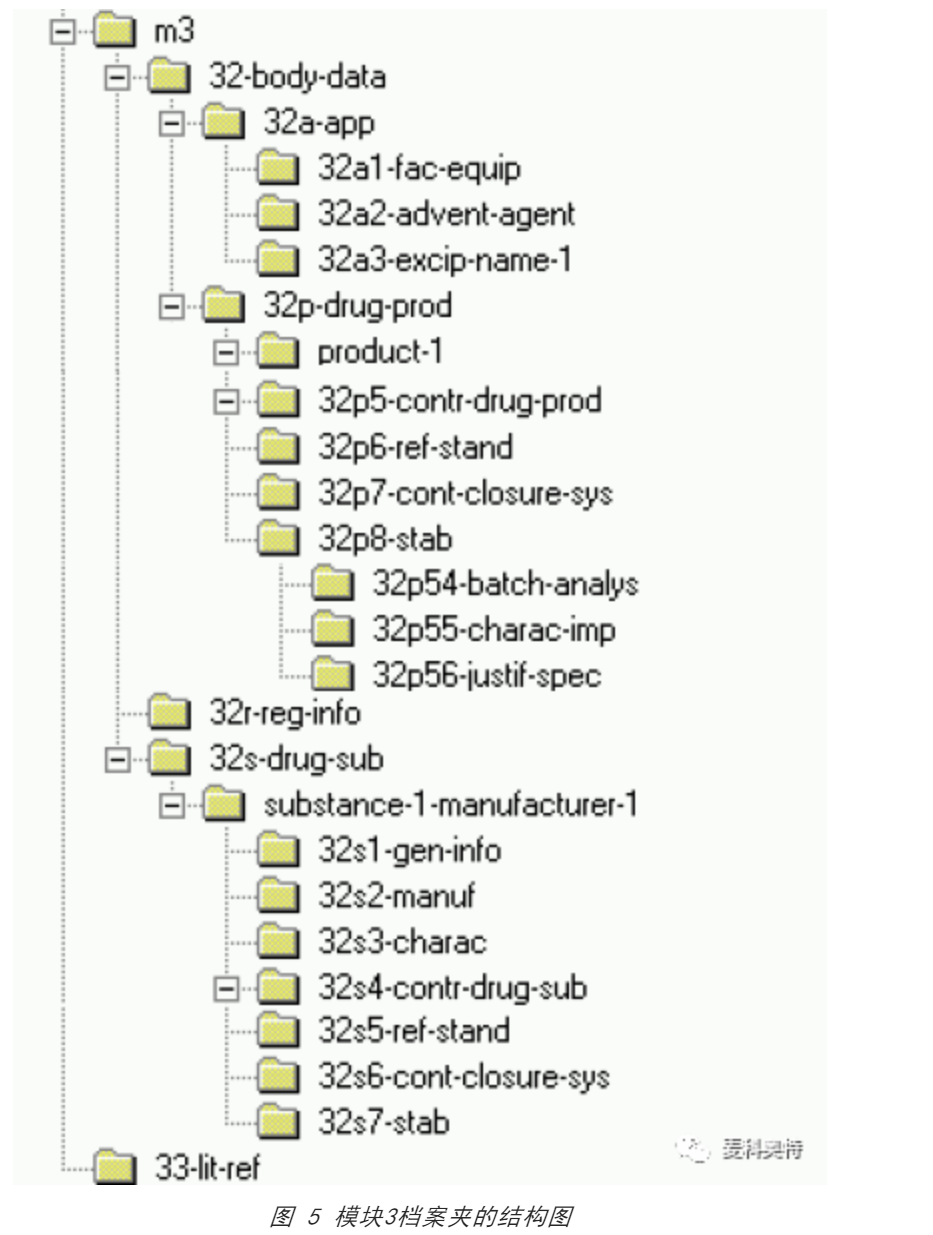
图 5 中的屏幕截图显示了模块 3 的代表性档案夹层结构及其次档案夹的名称。
模块 4 非临床研究报告:模块 4 的档案夹名称应为 m4,此模块最主要包含的文件为非临床的药理、药代与毒理的所有相关的实验报告。模块 4 中的档案夹应命名请看图 6,其次档案夹的名称可以进一步减少或以最小化档案夹名称来解决档案名称长度的问题。
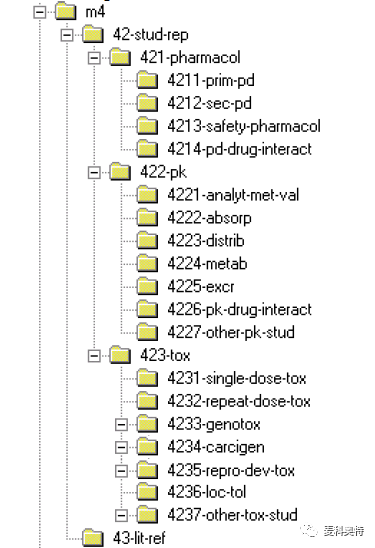
图 6 模块4档案夹的结构图
模块 5 临床研究报告:模块 5 的档案夹名称应为 m5,此模块最主要包含的文件为临床的报告。eCTD 组织在模块 5.3.7 中提供病例报告表和个体患者数据列表,在模块 5.4 中提供参考文献的所有文件。模块 4 中的档案夹应命名请看图 7,其次档案夹的名称可以进一步减少或以最小化档案夹名称来解决档案名称长度的问题。
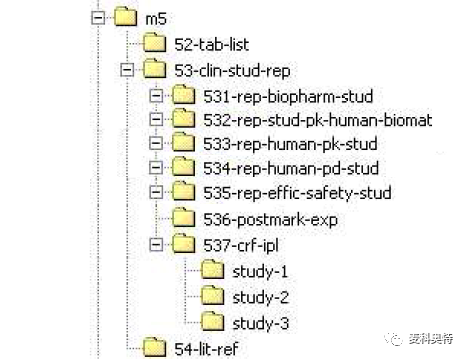
图 7 模块5档案夹的结构图
新型药物的eCTD申报全生命周期 (Lifecycle) 管理的流程包括三个层级: i) 临床试验/新药申请、ii) 首次/补充申请,和 iii) 补正/发补回复的序列 (图 8)。
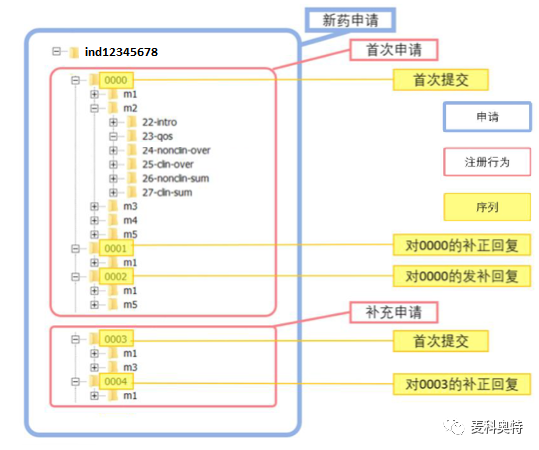
图 8 新型药物 ind12345678的eCTD申报全生命周期 (Lifecycle) 管理的模拟流程
eCTD 申报软件的选购
中国药企在中国和/或美国医事法规监管机构申报 eCTD新药申请,必须购买商业化的 eCTD 编辑软体,并训练相关注册人员后,才可输出所提交的全套申报资料,现在在中国可以选择的 eCTD软件并有其技术服务团队的产品有: i) Lorenz (益睿思), ii) Extedo (泰和镁科技), iii) Veeva, iv) GlobalSubmit, v) LIQUENT insight (Parexel), vi) eCTDXpress,ISIpublisher Reg tracker, ISItoolbox, TRS (CDC, Image Solutions, Inc., ISI), vii) eCTDmanager (创腾), viii) 柯里特Collietech eCTD, ix) BXeCTD (宝信软件),及 x) DoubleBridge (北京和桥)。
在购买eCTD申报软件应考虑下面的因素,必须考虑软件供应商是否可提供: a) 自动生成/辅助 PDF文件的目录 (Table of contents, TOC)、书签 (Bookmarks) 和超链接 (Hyperlinks), 若没有主动提供,是否可以跟其他辅助 eCTD 编辑程式互相配合的界面 (Interface), b) 与其他旧友的资料库是否有快速转移众多档案的界面,c) 与各公司的化学分析系统和其他临床资料互联,d) eCTD 认证/验证 (Validation) 软件,e) 24小时/每周7天的技术服务, f) 与医事法规监管机构最新的法规对于申报软件的更新能力, g) 合理的售价及每年维修的费用, h) 过去成功用于医事法规监管机构 (包括: NMPA, FDA 及 EMA) 申报经验,i) 提供优秀的 eCTD注册软件操作训练,及j) 人性化 eCTD 的操作模式及步骤。
人员的训练及申报文件的验证
如何使中国药企在高效下完成一套eCTD 申报? 根据过去的经验发现 - 药企必须经由优良的人员的训练方面才能得以完成,其中包括有: i) 三大部分 (即 CMC, 临床前及临床) 的 eCTD 申报人员的专业技能、明确的岗位职责、分工与衔接及优良的养成专业技能, ii) 在流程方面必须有良好的培训机制,有好质量的文件模板 (Templates)、完善的标准操作程序 (Standard operating procedures, SOPs)、顺畅的文件流转、有良好的审核流程,还有优良的项目管理, iii) 在 eCTD 平台上系统方面:要有好操作/维护的 eCTD 的软件及附属编辑软件 (e.g. Accenture's StartingPoint Submission Authoring Suite)、电子提交网关 (Electronic submissions gateway, ESG)、杀病毒 (Anti-virus) 软件、符合法规的电子签章 (e-signature)、光学字符识别系统 (Optical character recognition, OCR), 和Document management system (DMS) 系统,以上所谈及的项目必须互相相辅相成,才能有助于整个 eCTD 申报系统完美的演出。
任何药企项目的 eCTD全套申报资料都做好之后,必须通过官方的验证 (Validation) 才能递交成功。如果不能通过的话,申请者不得不重新把资料拿回来,在解决验证中出现的问题后,再次递交。若在新药申请递交过程中,发生这样的问题,势必会影响审评时限。所以药企必须在现有的eCTD 编辑软件上加装一套验证软件,资料递交之前先验证一遍,在通过官方验证的概率就会大大增加,为药企赢得宝贵的审评时限以及最大的商业利益。此外,中国药企一旦用了eCTD,后续的注册都需要用eCTD格式 (Once eCTD, always eCTD),因为 eCTD 工作量是巨大的,所以药企往往有两条路选择:i) 如果申报量少并且文件相对较少,可以外包给其他 eCTD publishing 技术服务的外包公司, 或 ii) 如果申报的是众多的新药研发项目加上大量的处理文件数量,后续众多项目必须与医事法规监管机构维护申请及补正式规章正/发补回复等相关事项,建议必须培养自己的电子注册以及医事法规处理团队,购买 eCTD 申报专业软件并训练自主自己专属的专业团队来做。
总之,本报告旨在介绍在 IND 注册的议题上,为中国药企建立 eCTD 管理系统提供意见,并提供购买eCTD申报软件应考虑因素,以及提供其发展所需的人员专业技能、岗位职责及人员培养。因为使用eCTD软件新药申报,其申报过程及管理是一个复杂繁琐的工程,此报告的分析为使用 eCTD 软件新药申报提供了一个通用框架,并突显 eCTD软件购买所需考虑的因素、申报人员训练及药企项目申报的需求大小规模做进一步的分析。本报告的另外一个目的是介绍 eCTD 软件申报流程及 eCTD 发展需求做范例,提供资讯给中国药企在中国/美国有意使用eCTD软件申报管理,以期了解用于支持其新型药物用 eCTD 申报模式,希望有助于国内药企在中国/美国 eCTD 注册的研究和策略的制定。
参考文献
Extedo eRegulatory Affairs, 2022, Taiwan FDA Chooses Extedo’s solutions for validation & reviewing @ https://www.extedo.com/company/news/detail/taiwan-fda-chooses-extedos-solutions-for-validation-reviewing
ICH guidance, M4 Organization of the Common Technical Document for the Registration of Pharmaceuticals for Human Use, Center for Drug uation and Research (CDER), U.S. FDA, October 2017 @ https://www.fda.gov/files/drugs/published/M4-Organization-of-the-Common-Technical-Document-for-the-Registration-of-Pharmaceuticals-for-Human-Use-Guidance-for-Industry.pdf
ICH eCTD specification V 3.2.2, ICH M2 EWG, Electronic common technical document specification 2008 @ https://admin.ich.org/sites/default/files/inline-files/eCTD_Specification_v3_2_2_0.pdf
卫生福利部食品药物管理署, 药品查验登记电子通用技术文件验证指引 (草案) 2020 @ https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&ved=2ahUKEwjn56P04aL3AhW2mFYBHc5GA84QFnoECCYQAQ&url=https%3A%2F%2Fregulation.cde.org.tw%2Fdata%2Fdownloadfile.php%3Fsid%3D1459&usg=AOvVaw0Rx_2IgawFmBrE0HD 0-Zc
药最网 医药注册研发资讯, eCTD注册资料准备中需要哪些专业软件?目前市场上主要有哪些公司提供这些软件?2017 @ https://www.yaozui.com/p/426701
申秀蕊 2022 药闻天下4月27了解 eCTD,看这篇文章就够了!@ https://zhiku.bopuyun.com/article/117304402779?zch=lvGAb9
Wikipedia 百科 2020 @ https://en.wikipedia.org/wiki/
免责声明: 如您(单位或个人)认为本文信息部分有侵权嫌疑,敬请立即通知我们,我们将在第一时间予以更正或删除。以上声明之解释权归本公司所有。谢谢。 |